This study examines the Twin-Arginine Translocase (Tat) system, especially the TatC subunit's role and variations between Gram-positive and Gram-negative bacteria. It investigates how hydrophobicity affects the Tat pathway, particularly in the interaction of the Escherichia coli (E. coli) TatC subunit and Bacillus substilis (B. subtilis) with SufI and TorA signal peptides. Different bioinformatics tools were used in the following research such as NCBI, Clustal Omega, MAFFT for sequence alignment, Phyre2 for structural modelling, and PyMOL, HDOCK, POCASA, KVFinder for protein docking and hydrophobicity analysis. The study provides an in-depth examination of TatC's structure, evolutionary relationships, and interactions with signal peptides. This approach uncovers the crucial balance between hydrophobic and hydrophilic forces in the Tat pathway, challenging the traditional emphasis on the twin-arginine motif in the SufI and TorA signal peptide. The analysis reveals the binding affinities and the pivotal role of the regions of the signal peptide interactions within TatC subunit in particular from Gram-negative E. coli and Gram-positive B. subtilis, enriching comprehension of the system's flexibility and the fundamental influence of hydrophobicity in protein interactions. The current study also demonstrates that peptides can bind effectively without twin-arginine motifs and suggests a deeper embedding of signal peptides in TatC's hydrophobic zones.
| Published in | Computational Biology and Bioinformatics (Volume 13, Issue 1) |
| DOI | 10.11648/j.cbb.20251301.13 |
| Page(s) | 22-41 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Twin Arginine Translocase, Protein Translocation Dynamics, Bioinformatics, Hydrophobic Interactions
Gram-Negative Bacteria | Classification | Accession | Amino Acids | Identity (%) |
|---|---|---|---|---|
Pseudomonas aeruginosa | Opportunistic Pathogen (Diggle & Whiteley, 2019) | PWU38561.1 | 267 | 57.60 |
Vibrio cholerae | Aquatic Pathogen (Halpern & Izhaki, 2017) | EGQ9206430.1 | 250 | 66.40 |
Salmonella enterica | Specialised invasive Pathogen (Hong et al., 2023) | EJJ4019649.1 | 259 | 91.12 |
Klebsiella pneumoniae | Respiratory Pathogen (Mannion et al., 2023) | CDO16241.1 | 259 | 83.72 |
Gram-Positive Bacteria | Classification | Accession | Amino Acids | Identity with TatCd (%) | Identity with TatCy (%) |
|---|---|---|---|---|---|
Bacillus cereus | Opportunistic Pathogen (Zheng et al., 2024) | HDR4908830 | 248 | 60.34 | 43.90 |
Paenibacillus sp. Tmac-D7 | Aquatic Pathogen (Sáez-Nieto et al., 2017) | WP_141336258.1 | 241 | 63.07 | 46.72 |
Streptococcus thermophilus | Specialised non-pathogen (Xu et al., 2023) | WP_084825977.1 | 242 | 41.95 | 36.21 |
Streptococcus pneumoniae | Respiratory Pathogen (Peng et al., 2023) | WP_050251502.1 | 243 | 36.71 | 35.27 |
Gram-negative Bacteria | Gram-positive Bacteria | Classification | Identity similarity (%) | |
|---|---|---|---|---|
Pseudomonas aeruginosa | Bacillus cereus | Opportunistic Pathogen | 28 | |
Vibrio cholerae | Paenibacillus sp. Tmac-D7 | Aquatic Pathogen | 28.57 | |
Salmonella enterica | Streptococcus thermophilus | Specialised Pathogen | 24.60 | |
Klebsiella pneumoniae | Streptococcus pneumoniae | Respiratory Pathogen | 27.20 | |
Escherichia coli str. K-12 | Bacillus subtilis | Non - pathogenic | TatCd 32.93 | TatCy 31.35 |
Protein Type | LGscore | MaxSub |
|---|---|---|
Receptor (TatC) | 2.676 | 0.162 |
Ligand (Signal Peptide) | 0.180 | -0.035 |
Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
Docking Score | -265.20 | -247.19 | -238.20 | -236.23 | -234.70 | -233.82 | -233.29 | -229.53 | -229.94 | -227.67 |
Confidence Score | 0.9092 | 0.8748 | 0.8537 | 0.8487 | 0.8447 | 0.8424 | 0.8410 | 0.8390 | 0.8319 | 0.8254 |
Ligand rmsd (Å) | 29.09 | 49.03 | 46.60 | 28.45 | 46.90 | 34.82 | 28.92 | 28.64 | 46.00 | 28.46 |
Protein Type | LGscore | MaxSub |
|---|---|---|
Receptor (TatC) | 2.676 | 0.162 |
Ligand (Signal Peptide) | 0.046 | -0.006 |
Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
Docking Score | -244.0 | -255.5 | -224.6 | -224.6 | -222.7 | -221.8 | -220.9 | -220.2 | -216.5 | -214.0 |
Confidence Score | 0.867 | 0.812 | 0.816 | 0.816 | 0.810 | 0.807 | 0.805 | 0.802 | 0.790 | 0.782 |
Ligand rmsd (Å) | 502.6 | 500.1 | 503.1 | 486.0 | 504.5 | 480.6 | 501.3 | 479.0 | 502.1 | 479.9 |
Protein Type | LGscore | MaxSub |
|---|---|---|
Receptor (TatC) | 3.109 | 0.206 |
Ligand (Signal Peptide) | 0.534 | 0.056 |
Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
Docking Score | -244.7 | -227.6 | -222.9 | -222.2 | -219.6 | -218.4 | -217.1 | -213.6 | -210.9 | -210.9 |
Confidence Score | 0.869 | 0.825 | 0.811 | 0.809 | 0.801 | 0.797 | 0.792 | 0.781 | 0.772 | 0.771 |
Ligand rmsd (Å) | 488 | 504.4 | 503.6 | 488.0 | 503.9 | 502.9 | 503.7 | 503 | 502 | 510.6 |
Gram-Negative Bacteria | Pocket Binding Rank | Pocket Number | Pocket Volume Å | Average Volume -Depth value Å | Total Binding Sites Probe radius of 5Å |
|---|---|---|---|---|---|
Escherichia coli str. K-12 | 1 | 93 | 407 | 1108 | 15 |
2 | 28 | 231 | 724 | ||
3 | 162 | 254 | 720 | ||
4 | 265 | 114 | 288 | ||
5 | 123 | 99 | 239 | ||
Pseudomonas aeruginosa | 1 | 114 | 353 | 990 | 12 |
2 | 29 | 287 | 858 | ||
3 | 142 | 321 | 854 | ||
4 | 194 | 153 | 375 | ||
5 | 18 | 129 | 337 | ||
Vibrio cholerae | 1 | 147 | 456 | 1371 | 15 |
2 | 157 | 296 | 753 | ||
3 | 31 | 170 | 526 | ||
4 | 527 | 149 | 368 | ||
5 | 137 | 128 | 328 | ||
Salmonella enterica | 1 | 116 | 396 | 1079 | 16 |
2 | 159 | 254 | 720 | ||
3 | 28 | 216 | 646 | ||
4 | 258 | 114 | 288 | ||
5 | 124 | 78 | 190 | ||
Klebsiella pneumoniae | 1 | 127 | 596 | 1648 | 13 |
2 | 62 | 122 | 422 | ||
3 | 122 | 137 | 333 | ||
4 | 367 | 109 | 277 | ||
5 | 432 | 88 | 234 |
Gram-Positive Bacteria | Pocket Binding Rank | Pocket Number | Pocket Volume Å | Average Volume -Depth value Å | Total Binding Sites Probe radius of 5Å |
|---|---|---|---|---|---|
Bacillus subtilis TatCd / TatCy | 1 | 132 / 45 | 356 / 301 | 1059 / 866 | 12 / 17 |
2 | 201 / 193 | 188 / 297 | 507 / 846 | ||
3 | 52 / 126 | 109 / 264 | 371 / 680 | ||
4 | 189 / 342 | 138 / 235 | 343 / 575 | ||
5 | 263 / 93 | 129 / 62 | 320 / 161 | ||
Bacillus cereus | 1 | 163 | 392 | 1094 | 12 |
2 | 114 | 203 | 564 | ||
3 | 61 | 140 | 507 | ||
4 | 166 | 194 | 471 | ||
5 | 243 | 174 | 423 | ||
Paenibacillus | 1 | 343 | 656 | 1726 | 11 |
2 | 65 | 250 | 706 | ||
3 | 161 | 213 | 651 | ||
4 | 17 | 39 | 122 | ||
5 | 92 | 20 | 120 | ||
Streptococcus thermophilus | 1 | 293 | 635 | 1652 | 11 |
2 | 127 | 229 | 596 | ||
3 | 39 | 150 | 464 | ||
4 | 213 | 76 | 232 | ||
5 | 419 | 71 | 183 | ||
Streptococcus pneumoniae | 1 | 302 | 462 | 1206 | 10 |
2 | 43 | 219 | 717 | ||
3 | 113 | 233 | 638 | ||
4 | 129 | 77 | 204 | ||
5 | 358 | 64 | 184 |
| [1] | Alami, M., Lüke, I., Deitermann, S., Eisner, G., Koch, H.-G., Brunner, J., & Müller, M. (2003). Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Molecular Cell, 12(4), 937–946. |
| [2] | Blümmel, A.-S., Drepper, F., Knapp, B., Eimer, E., Warscheid, B., Müller, M., & Fröbel, J. (2017). Structural features of the TatC membrane protein that determine docking and insertion of a twin-arginine signal peptide. Journal of Biological Chemistry, 292(52), 21320–21329. |
| [3] | Bronstein, P., Marrichi, M., & DeLisa, M. P. (2004). Dissecting the twin-arginine translocation pathway using genome-wide analysis. Research in Microbiology, 155(10), 803–810. |
| [4] | Buck, E. D., Lammertyn, E., & Anné, J. (2008). The importance of the twin-arginine translocation pathway for bacterial virulence. Trends in Microbiology, 16(9), 442–453. |
| [5] | Buchanan, G., de Leeuw, E., Stanley, N. R., Wexler, M., Berks, B. C., Sargent, F., and Palmer, T. (2002) ‘Functional Complexity of the Twin-Arginine Translocase TatC Component Revealed by Site-Directed Mutagenesis.’ Molecular Microbiology [online] 43(6), 1457–70. available from |
| [6] | Capra, J. A., & Singh, M. (2007). Predicting functionally important residues from sequence conservation. Bioinformatics, 23(15), 1875–1882. |
| [7] | Castro-Alvarez, A., Costa, A., & Vilarrasa, J. (2017). The Performance of Several Docking Programs at Reproducing Protein–Macrolide-Like Crystal Structures. Molecules, 22(1), 136. |
| [8] | Ciemny, M. P., Debinski, A., Paczkowska, M., Kolinski, A., Kurcinski, M., & Kmiecik, S. (2016). Protein-peptide molecular docking with large-scale conformational changes: the p53-MDM2 interaction. Scientific Reports, 6(1). |
| [9] | Cline, K. (2015) ‘Mechanistic Aspects of Folded Protein Transport by the Twin Arginine Translocase (Tat).’ The Journal of Biological Chemistry [online] 290(27), 16530–8. |
| [10] | Dalbey, R. E. and Kuhn, A. (2012) ‘Protein Traffic in Gram-Negative Bacteria - How Exported and Secreted Proteins Find Their Way’. in FEMS Microbiology Reviews. vol. 36 (6). Oxford Academic, 1023–1045. |
| [11] | Diggle, S. P., & Whiteley, M. (2019). Microbe Profile: Pseudomonas aeruginosa: Opportunistic Pathogen and Lab Rat. Microbiology, 166(1), 30–33. |
| [12] | Dionisio, F., Domingues, C. P. F., Rebelo, J. S., Monteiro, F., & Nogueira, T. (2023). The Impact of Non-Pathogenic Bacteria on the Spread of Virulence and Resistance Genes. International Journal of Molecular Sciences, 24(3), 1967. |
| [13] | Fitzgerald, J. Ross., & Musser, J. M. (2001). Evolutionary genomics of pathogenic bacteria. Trends in Microbiology, 9(11), 547–553. |
| [14] | Frain, K. M., Robinson, C., & van Dijl, J. M. (2019). Transport of Folded Proteins by the Tat System. The Protein Journal, 38(4), 377–388. |
| [15] | Fröbel, J., Rose, P., & Müller, M. (2012). Twin-arginine-dependent translocation of folded proteins. Philosophical Transactions of the Royal Society B: Biological Sciences, 367(1592), 1029–1046. |
| [16] | Gérard, F., & Cline, K. (2006). Efficient Twin Arginine Translocation (Tat) Pathway Transport of a Precursor Protein Covalently Anchored to Its Initial cpTatC Binding Site. Journal of Biological Chemistry, 281(10), 6130–6135. |
| [17] | Ghosh, P., & Sowdhamini, R. (2017). Bioinformatics comparisons of RNA-binding proteins of pathogenic and non-pathogenic Escherichia coli strains reveal novel virulence factors. BMC Genomics, 18(1). |
| [18] | Guerra, J. V. D. S., Ribeiro Filho, H. V., Bortot, L. O., Honorato, R. V., Pereira, J. G. D. C., & Lopes-de-Oliveira, P. S. (2020). ParKVFinder: a thread-level parallel approach in biomolecular cavity detection. SoftwareX, 12, 100606. |
| [19] | Guerra, J. V. D. S., Ribeiro-Filho, H. V., Jara, G. E., Bortot, L. O., Pereira, J. G. D. C., & Lopes-de-Oliveira, P. S. (2021). pyKVFinder: an efficient and integrable python package for biomolecular cavity detection and characterization in data science. BMC Bioinformatics. |
| [20] | Guerra, J. V. S., Ribeiro-Filho, H. V., Pereira, J. G. C., & Lopes-de-Oliveira, P. S. (2023). KVFinder-web: a web-based application for detecting and characterizing biomolecular cavities. Nucleic Acids Research. |
| [21] | Habersetzer, J., Moore, K., Cherry, J., Buchanan, G., Stansfeld, P. J. and Palmer, T. (2017). Substrate-triggered position switching of TatA and TatB during Tat transport in Escherichia coli. Open Biology, 7(8), pp. 170091–170091. |
| [22] | Haliloglu, T., & Bahar, I. (2015). Adaptability of protein structures to enable functional interactions and evolutionary implications. Current Opinion in Structural Biology, 35, 17–23. |
| [23] | Halpern, M., & Izhaki, I. (2017). Fish as Hosts of Vibrio cholerae. Frontiers in Microbiology, 8. |
| [24] | HDOCK Group. (2024). HDOCK server for protein-protein and protein-DNA/RNA docking. Retrieved from |
| [25] | Hong, Y., Wu, Y., Xie, Y., Ben, L., Bu, X., Pan, X., Shao, J., Dong, Q., Qin, X., & Wang, X. (2023). Effects of antibiotic-induced resistance on the growth, survival ability and virulence of Salmonella enterica. Food Microbiology, 115, 104331–104331. |
| [26] | Hsin Liu, C., Li, K.-C., & Yuan, S. (2012). Human protein–protein interaction prediction by a novel sequence-based co-evolution method: co-evolutionary divergence. Bioinformatics, 29(1), 92–98. |
| [27] | Huang, Q., & Palmer, T. (2017). Signal Peptide Hydrophobicity Modulates Interaction with the Twin-Arginine Translocase. MBio, 8(4). |
| [28] | Kadeřábková, N., Furniss, C. D., Mahmood, A., & Mavridou, D. (2023). Making a chink in their armor: Current and next-generation antimicrobial strategies against the bacterial cell envelope. Advances in Microbial Physiology, 221–307. |
| [29] | Katoh, K., Rozewicki, J., & Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics, 20(4). |
| [30] | Katoh, K., & Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution, 30(4), 772–780. |
| [31] | Kawabata, T., & Go, N. (2007). Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins: Structure, Function, and Bioinformatics, 68(2), 516–529. |
| [32] | Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., & Sternberg, M. J. E. (2015). The Phyre2 Web Portal for Protein modeling, Prediction and Analysis. Nature Protocols, 10(6), 845–858. |
| [33] | Kuraku, S., Zmasek, C. M., Nishimura, O., & Katoh, K. (2013). aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Research, 41 (W1), W22–W28. |
| [34] | Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T., & Ben-Tal, N. (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Research, 33 (Web Server), W299–W302. |
| [35] | Lee, P. A., Tullman-Ercek, D., & Georgiou, G. (2006). The Bacterial Twin-Arginine Translocation Pathway. Annual Review of Microbiology, 60(1), 373–395. |
| [36] | Lu, H., Zhou, Q., He, J., Jiang, Z., Peng, C., Tong, R., & Shi, J. (2020). Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduction and Targeted Therapy, 5(1). |
| [37] | Madeira, F., Pearce, M., Tivey, A. R. N., Basutkar, P., Lee, J., Edbali, O., Madhusoodanan, N., Kolesnikov, A., & Lopez, R. (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Research, 50 (W1). |
| [38] | Mannion, J. M., Segal, B. M., McLoughlin, R. M., & Lalor, S. J. (2023). Respiratory tract Moraxella catarrhalis and Klebsiella pneumoniae can promote pathogenicity of myelin-reactive Th17 cells. Mucosal Immunology. |
| [39] | Mehner-Breitfeld, D., Ringel, M. T., Tichy, D. A., Endter, L. J., Stroh, K. S., Heinrich Lünsdorf, Herre Jelger Risselada, & Brüser, T. (2022). TatA and TatB generate a hydrophobic mismatch important for the function and assembly of the Tat translocon in Escherichia coli. Journal of Biological Chemistry, 298(9), 102236–102236. |
| [40] | Mendel, S., McCarthy, A., Barnett, J. P., Eijlander, R. T., Nenninger, A., Kuipers, O. P., & Robinson, C. (2008). The Escherichia coli TatABC System and a Bacillus subtilis TatAC-type System Recognise Three Distinct Targeting Determinants in Twin-arginine Signal Peptides. Journal of Molecular Biology, 375(3), 661–672. |
| [41] | National Center for Biotechnology Information (NCBI). (2024). BLAST: Basic Local Alignment Search Tool. Retrieved [January, 2024], from |
| [42] | Oba, M., Nakajima, S., Kurumi Misao, Hidetomo Yokoo, & Tanaka, M. (2023). Effect of helicity and hydrophobicity on cell-penetrating ability of arginine-rich peptides. Bioorganic & Medicinal Chemistry, 91, 117409–117409. |
| [43] | Palmer, T., & Stansfeld, P. J. (2020). Targeting of proteins to the twin‐arginine translocation pathway. Molecular Microbiology, 113(5), 861–871. |
| [44] | Pei, D., & Dalbey, R. E. (2022). Membrane translocation of folded proteins. Journal of Biological Chemistry, 298(7), 102107. |
| [45] | Peng, S., Guo, C., Cui, H., & Duan, Z. (2023). Complete genome analysis of Lactiplantibacillus plantarum VHProbi P06, a novel probiotic that resists Streptococcus pneumoniae in the upper respiratory tract. International Journal of Biological Macromolecules, 253, 127320–127320. |
| [46] | Potteth, U. S., Upadhyay, T., Saini, S., & Ishu Saraogi. (2021). Novel Antibacterial Targets in Protein Biogenesis Pathways. ChemBioChem, 23(4). |
| [47] | Rollauer, S. E., Tarry, M. J., Graham, J. E., Jääskeläinen, M., Jäger, F., Johnson, S., Krehenbrink, M., Liu, S.-M., Lukey, M. J., Marcoux, J., McDowell, M. A., Rodriguez, F., Roversi, P., Stansfeld, P. J., Robinson, C. V., Sansom, M. S. P., Palmer, T., Högbom, M., Berks, B. C., and Lea, S. M. (2012) ‘Structure of the TatC Core of the Twin-Arginine Protein Transport System’. Nature [online] 492(7428), 210–214. available from |
| [48] | Ren, A., Ishida, T., & Akiyama, Y. (2013). Assessing statistical reliability of phylogenetic trees via a speedy double bootstrap method. Molecular Phylogenetics and Evolution, 67(2), 429–435. |
| [49] | Ribeiro, A. A. S. T., Horta, B. A. C., & de Alencastro, R. B. (2021). KVFINDER (v1.2.0) [Software]. Available from |
| [50] | Robinson, S. W., Afzal, A. M., & Leader, D. P. (2014). Bioinformatics: Concepts, Methods, and Data. Handbook of Pharmacogenomics and Stratified Medicine, 259–287. |
| [51] | Sáez-Nieto, J. A., Medina-Pascual, M. J., Carrasco, G., Garrido, N., Fernandez-Torres, M. A., Villalón, P., & Valdezate, S. (2017). Paenibacillus spp. isolated from human and environmental samples in Spain: detection of 11 new species. New Microbes and New Infections, 19, 19–27. |
| [52] | Schrödinger, LLC. (2024). PyMOL (Ver. 2.0) [Webserver]. Available from |
| [53] | Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., & Higgins, D. G. (2014). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 7(1), 539. |
| [54] | Tara, Tran, V. A., & Turner, R. J. (2013). The Hydrophobic Region of the DmsA Twin-Arginine Leader Peptide Determines Specificity with Chaperone DmsD. Biochemistry, 52(43), 7532–7541. |
| [55] | Tarry, M. J., Schäfer, E., Chen, S., Buchanan, G., Greene, N. P., Lea, S. M., Palmer, T., Saibil, H. R., and Berks, B. C. (2009) ‘Structural Analysis of Substrate Binding by the TatBC Component of the Twin-Arginine Protein Transport System.’ Proceedings of the National Academy of Sciences of the United States of America [online] 106(32), 13284–9. |
| [56] | Taw, M. N., Li, M., Kim, D., Rocco, M. A., DeLisa, M. P., & Zhmayev, D. (2021). Engineering a Supersecreting Strain of Escherichia coli by Directed Coevolution of the Multiprotein Tat Translocation Machinery. ACS Synthetic Biology, 10(11), 2947–2958. |
| [57] | Ulfig, A., Fröbel, J., Lausberg, F., Blümmel, A.-S., Heide, A. K., Müller, M., & Freudl, R. (2017). The h-region of twin-arginine signal peptides supports productive binding of bacterial Tat precursor proteins to the TatBC receptor complex. Journal of Biological Chemistry, 292(26), 10865–10882. |
| [58] | Ulfig, A., & Freudl, R. (2018). The early mature part of bacterial twin-arginine translocation (Tat) precursor proteins contributes to TatBC receptor binding. Journal of Biological Chemistry, 293(19), 7281–7299. |
| [59] | Uniprot. (2023). UniProt. Uniprot.org. |
| [60] | Voulhoux, R. (2001). Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. The EMBO Journal, 20(23), 6735–6741. |
| [61] | Widdick, D. A., Dilks, K., Chandra, G., Bottrill, A., Naldrett, M., Pohlschroder, M., & Palmer, T. (2006). The twin-arginine translocation pathway is a major route of protein export in Streptomyces coelicolor. Proceedings of the National Academy of Sciences, 103(47), 17927–17932. |
| [62] | Wingert, B., Krieger, J., Li, H., & Bahar, I. (2021). Adaptability and specificity: how do proteins balance opposing needs to achieve function? Current Opinion in Structural Biology, 67, 25–32. |
| [63] | Xu, Y., Hu, J., Liu, D., Tang, J., Liang, M., Wu, J., & Xiong, J. (2023). Assessment of the safety and metabolism characteristics of Streptococcus thermophilus DMST-H2 based on complete genome and phenotype analysis. LWT, 184, 114907–114907. |
| [64] | Yan, Y., Tao, H., He, J., & Huang, S.-Y. (2020). The HDOCK server for integrated protein-protein docking. Nature Protocols. |
| [65] | Yu, J., Zhou, Y., Tanaka, I., & Yao, M. (2010). Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics, 26(1), 46-52. |
| [66] | Zhang, Y., Wang, L., Hu, Y., & Jin, C. (2014). Solution structure of the TatB component of the twin-arginine translocation system. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1838(7), 1881–1888. |
APA Style
Correia, M. S., Williams, S. M. (2025). Comparative Study of the Twin Arginine Translocase (Tat) System Across Bacterial Species: Insights into Hydrophobic Interactions, Signal Peptide Binding and Protein Translocation Dynamics. Computational Biology and Bioinformatics, 13(1), 22-41. https://doi.org/10.11648/j.cbb.20251301.13
ACS Style
Correia, M. S.; Williams, S. M. Comparative Study of the Twin Arginine Translocase (Tat) System Across Bacterial Species: Insights into Hydrophobic Interactions, Signal Peptide Binding and Protein Translocation Dynamics. Comput. Biol. Bioinform. 2025, 13(1), 22-41. doi: 10.11648/j.cbb.20251301.13
AMA Style
Correia MS, Williams SM. Comparative Study of the Twin Arginine Translocase (Tat) System Across Bacterial Species: Insights into Hydrophobic Interactions, Signal Peptide Binding and Protein Translocation Dynamics. Comput Biol Bioinform. 2025;13(1):22-41. doi: 10.11648/j.cbb.20251301.13
@article{10.11648/j.cbb.20251301.13,
author = {Micael Sousa Correia and Sharon Mendel Williams},
title = {Comparative Study of the Twin Arginine Translocase (Tat) System Across Bacterial Species: Insights into Hydrophobic Interactions, Signal Peptide Binding and Protein Translocation Dynamics
},
journal = {Computational Biology and Bioinformatics},
volume = {13},
number = {1},
pages = {22-41},
doi = {10.11648/j.cbb.20251301.13},
url = {https://doi.org/10.11648/j.cbb.20251301.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cbb.20251301.13},
abstract = {This study examines the Twin-Arginine Translocase (Tat) system, especially the TatC subunit's role and variations between Gram-positive and Gram-negative bacteria. It investigates how hydrophobicity affects the Tat pathway, particularly in the interaction of the Escherichia coli (E. coli) TatC subunit and Bacillus substilis (B. subtilis) with SufI and TorA signal peptides. Different bioinformatics tools were used in the following research such as NCBI, Clustal Omega, MAFFT for sequence alignment, Phyre2 for structural modelling, and PyMOL, HDOCK, POCASA, KVFinder for protein docking and hydrophobicity analysis. The study provides an in-depth examination of TatC's structure, evolutionary relationships, and interactions with signal peptides. This approach uncovers the crucial balance between hydrophobic and hydrophilic forces in the Tat pathway, challenging the traditional emphasis on the twin-arginine motif in the SufI and TorA signal peptide. The analysis reveals the binding affinities and the pivotal role of the regions of the signal peptide interactions within TatC subunit in particular from Gram-negative E. coli and Gram-positive B. subtilis, enriching comprehension of the system's flexibility and the fundamental influence of hydrophobicity in protein interactions. The current study also demonstrates that peptides can bind effectively without twin-arginine motifs and suggests a deeper embedding of signal peptides in TatC's hydrophobic zones.},
year = {2025}
}
TY - JOUR T1 - Comparative Study of the Twin Arginine Translocase (Tat) System Across Bacterial Species: Insights into Hydrophobic Interactions, Signal Peptide Binding and Protein Translocation Dynamics AU - Micael Sousa Correia AU - Sharon Mendel Williams Y1 - 2025/07/22 PY - 2025 N1 - https://doi.org/10.11648/j.cbb.20251301.13 DO - 10.11648/j.cbb.20251301.13 T2 - Computational Biology and Bioinformatics JF - Computational Biology and Bioinformatics JO - Computational Biology and Bioinformatics SP - 22 EP - 41 PB - Science Publishing Group SN - 2330-8281 UR - https://doi.org/10.11648/j.cbb.20251301.13 AB - This study examines the Twin-Arginine Translocase (Tat) system, especially the TatC subunit's role and variations between Gram-positive and Gram-negative bacteria. It investigates how hydrophobicity affects the Tat pathway, particularly in the interaction of the Escherichia coli (E. coli) TatC subunit and Bacillus substilis (B. subtilis) with SufI and TorA signal peptides. Different bioinformatics tools were used in the following research such as NCBI, Clustal Omega, MAFFT for sequence alignment, Phyre2 for structural modelling, and PyMOL, HDOCK, POCASA, KVFinder for protein docking and hydrophobicity analysis. The study provides an in-depth examination of TatC's structure, evolutionary relationships, and interactions with signal peptides. This approach uncovers the crucial balance between hydrophobic and hydrophilic forces in the Tat pathway, challenging the traditional emphasis on the twin-arginine motif in the SufI and TorA signal peptide. The analysis reveals the binding affinities and the pivotal role of the regions of the signal peptide interactions within TatC subunit in particular from Gram-negative E. coli and Gram-positive B. subtilis, enriching comprehension of the system's flexibility and the fundamental influence of hydrophobicity in protein interactions. The current study also demonstrates that peptides can bind effectively without twin-arginine motifs and suggests a deeper embedding of signal peptides in TatC's hydrophobic zones. VL - 13 IS - 1 ER -
College of Engineering, Environment and Science, School of Science, Coventry University, Coventry, UK
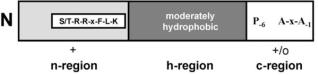
Figure 1. Signal Peptide SufI regions: Tat Signal Sequence, image provided by Bronstein et al., 2004 [3], presents the n-region with the S/T-RR-x-F-L-K motif, followed by the moderately hydrophobic h-region, and the c-region containing the A-x-A amino acid sequence.
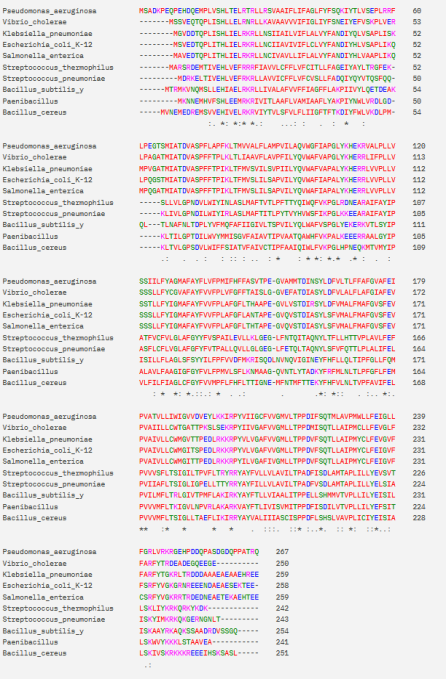
Figure 2. Gram-negative and Gram-positive Multiple Sequence Alignment of TatC subunit.
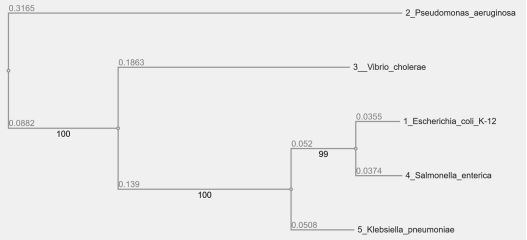
Figure 3. Gram-negative Bacteria Phylogenetic Tree.
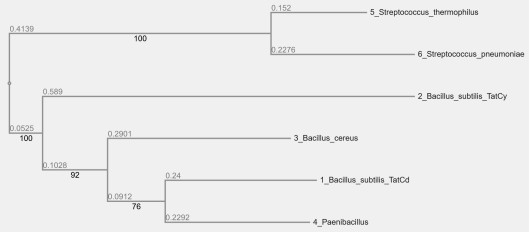
Figure 4. Gram-positive Bacteria Phylogenetic Tree.
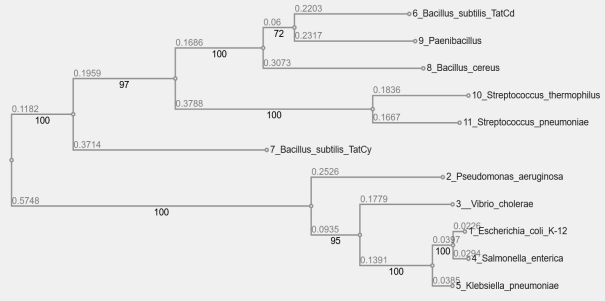
Figure 5. Gram-negative and Gram-positive Bacteria Phylogenetic Tree.
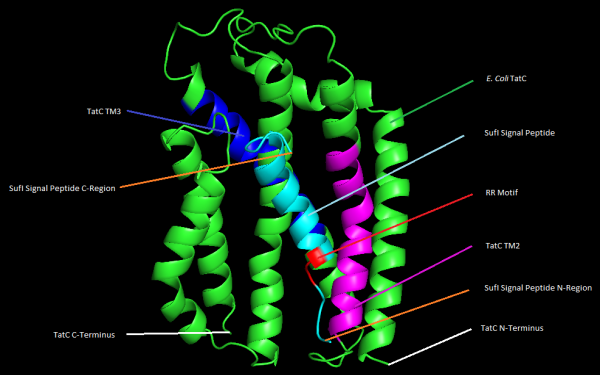
Figure 6. 3D representation of E. coli str. K-12 TatC and SufI protein-protein docking with highlighted features.
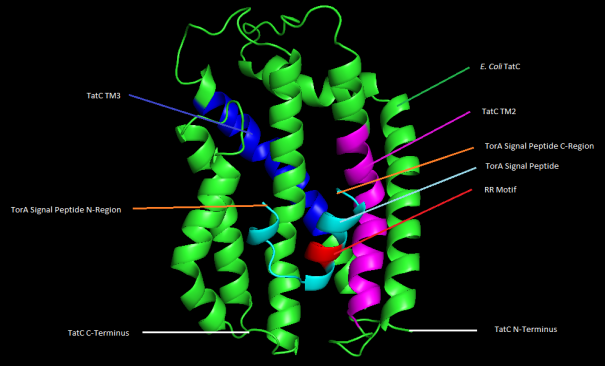
Figure 7. 3D representation of E. coli str. K-12 TatC and TorA protein-protein docking with highlighted features.
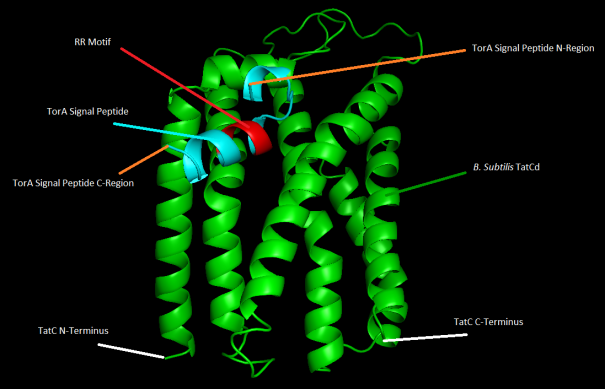
Figure 8. 3D representation of B. subtilis TatCd and TorA protein-protein docking with highlighted features.
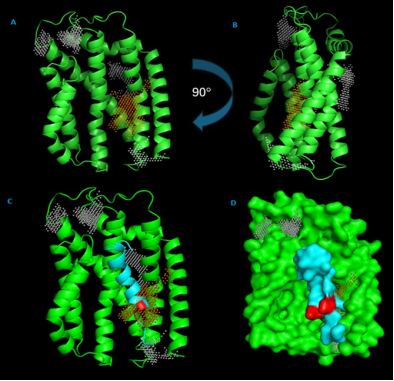
Figure 9. Structural Analysis of E. coli str. K-12 TatC with Binding Sites and Motifs.
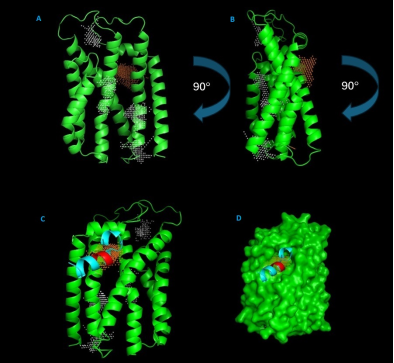
Figure 10. Structural Analysis of B. subtilis TatCd with Binding Sites and Motifs.
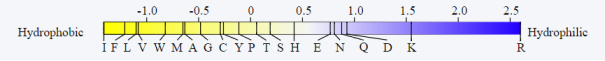
Figure 11. KVFinder Hydrophobicity Metrics.
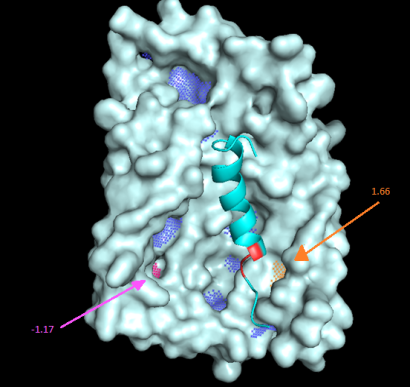
Figure 12. Hydrophobicity Mapping in E. coli str. K-12 TatC Protein.
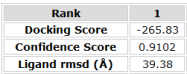
Figure 13. Results from the modified SufI Sequence that allocates the double Isoleucines.
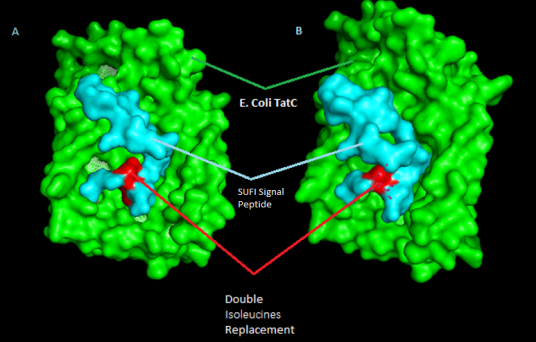
Figure 14. E. coli str. K-12 TatC docking with SufI mutated sequence.
Information

