This theoretical work give a comprehensive properties of a Schiff base compound that is formed when amines and ketone or aldehyde are combined. The investigated Schiff base compounds were designed by combining primary amines with ketones or aldehydes. The spectroscopic characteristics, molecular structure, electrostatic potential maps, and other molecular properties of these compounds had been computed at the B3LYP Functional with a 6-311G(d,p) basic set. The optimization and transition states of the molecules were analyzed by applying the B3LYP Functional with a 6-31G(d,p) basic set, based on density functional theory (DFT) and time-dependent density functional theory (TD-DFT) for ground-state and excited-state calculations, respectively. We had also determine the band length and band angles of the designed molecules. Several computational software packages was used to study the spectroscopic, electronics, and molecular characteristics of explore Schiff base compounds. The molecules were designed in GaussView5 and optimized in Gaussian 09. PyMolyze and Origin6.0 software were used to perform the density of state (DOS) analysis & to draw the absorption spectra of probe molecules. TDM analysis were conducted to determine the charge distribution in the investigated molecules using Multiwfn3.7 and VMD1.9.1 software. Correlation statistical models were employed to interpret the statistical data. The docking results of the designed molecules were compared with antibacterial standards, and we expect these results to show greater efficiency than the reference molecules. Additionally, the antitumor and antibacterial characteristics of a designed molecule were compared with those of the reference molecules.
| Published in | International Journal of Computational and Theoretical Chemistry (Volume 14, Issue 1) |
| DOI | 10.11648/j.ijctc.20261401.12 |
| Page(s) | 15-33 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
DFT, B3LYP, Schiff Base, Optimization, Transition States, Spectroscopic
Molecules | Bonds | Vibrational Modes | Frequency (cm-1) | Intensity |
|---|---|---|---|---|
SL | C—N | Scissoring | 809.06 | 600.34 |
H—N | Symmetrical | 1204.39 | 300 | |
O—N | Symmetrical | 1425.63 | 3500 | |
SL1 | H—O—N | Scissoring | 610.05 | 1521.62 |
H—N | Scissoring | 809.44 | 964.23 | |
C—N | Scissoring | 825.42 | 703.75 | |
O—N | Symmetrical | 1063.32 | 589.39 | |
N—C—O | Anti-Symmetrical | 1144.25 | 2813.24 | |
O—N—O | Anti-Symmetrical | 1332.40 | 1717.22 | |
N—C—N | Anti-Symmetrical | 1397.71 | 2864.40 | |
C—N—N | Anti-Symmetrical | 1421.33 | 3423.06 | |
N—C—H | Anti-Symmetrical | 1439.72 | 859.32 | |
H—N—O | Anti-Symmetrical | 1631.41 | 1146.36 | |
SL2 | O—C—H | Symmetrical | 395.88 | 1308.31 |
N—O—H | Symmetrical | 611.14 | 1267.08 | |
H—N—C | Symmetrical | 822.71 | 1335.77 | |
N—O—H | Anti-Symmetrical | 887.12 | 843.42 | |
H—N—O | Anti-Symmetrical | 1137.45 | 2965.11 | |
H—N—N | Anti-Symmetrical | 1388.37 | 4391.08 | |
N—C—N | Anti-Symmetrical | 1412.25 | 5636.42 | |
SL3 | Cl—O—H | Rocking | 420.27 | 1041.28 |
O—C -O | Anti-Symmetrical | 578.42 | 1147.82 | |
H—O -H | Symmetrical | 613.47 | 1080.07 | |
H—C—N | Scissoring | 1126.47 | 4517.31 | |
C—N—Cl | Anti-Symmetrical | 1400.46 | 5476.00 | |
N—C -N | Anti-Symmetrical | 1416.62 | 3148.02 |
Molecules | Bonds | Vibrational Modes | Frequency | Raman | Intensity |
|---|---|---|---|---|---|
SL | C—N—H | Scissoring | 327.13 | 5.2388 | 1.3 |
N—C—N | Scissoring | 380.49 | 7.7635 | 1.2 | |
H—N—H | Scissoring | 562.75 | 0.0346 | 4.4 | |
C—O—N | Wagging | 754.41 | 2.3969 | 0.4 | |
H—N—C | Scissoring | 807.17 | 3.6844 | 0.3 | |
N—C—H | Wagging | 881.15 | 13.1493 | 1 | |
C—C—N | Symmetric | 1051.63 | 0.9426 | 0.2 | |
H—N—H | Anti-Symmetric | 1183.52 | 2.3888 | 0.4 | |
H—N—C | Anti-Symmetric | 1318.24 | 2.7109 | 0.2 | |
C—N—BH | Symmetric | 1447.19 | 1.3304 | 0.2 | |
SL1 | O—N—O | Twisting | 31.98 | 1.2865 | 0.1 |
H—O—C | Scissoring | 300.87 | 5.9279 | 0.3 | |
C—N—H | Scissoring | 345.00 | 6.3461 | 0.4 | |
N—C—N | Scissoring | 384.16 | 3.7635 | 0.2 | |
O—C—O | Anti-Symmetric | 567.28 | 5.1622 | 0.4 | |
C—N—H | Twisting | 631.71 | 18.5154 | 1.1 | |
B—Li—O | Symmetric | 708.17 | 19.5818 | 1.2 | |
N—O—N | Symmetric | 873.28 | 12.684 | 0.6 | |
O—N—O | Symmetric | 1063.32 | 25.4996 | 1.5 | |
H—N—C | Anti-Symmetric | 1318.32 | 3.7423 | 0.2 | |
N—N—O | Anti-Symmetric | 1417.23 | 4.1945 | 0.2 | |
H—N—O | Anti-Symmetric | 1631.41 | 8.7024 | 0.5 | |
SL2 | H—N—O | Twisting | 19.41 | 2.1398 | 0.2 |
O—N—H | Scissoring | 284.95 | 4.8997 | 0.4 | |
N—O—H | Wagging | 308.07 | 0.5315 | 0.4 | |
H—O—C | Symmetric | 445.85 | 4.9158 | 0.4 | |
C—N—O | Anti-Symmetric | 577.36 | 28.8333 | 2.3 | |
C—N—N | Scissoring | 789.73 | 12.5613 | 0.9 | |
H—C—N | Wagging | 867.20 | 15.5730 | 1.2 | |
O—N—C | Symmetric | 1098.43 | 44.5076 | 3.4 | |
C—O—H | Anti-Symmetric | 1290.31 | 8.0811 | 0.7 | |
O—N—C | Anti-Symmetric | 1351.23 | 1.4670 | 0.3 | |
H—C—N | Anti-Symmetric | 1451.62 | 7.6134 | 0.7 | |
O—N—C | Anti-Symmetric | 1611.33 | 15.2449 | 1.3 | |
SL3 | C—O—C | Scissoring | 303.21 | 4.4150 | 0.3 |
N—O—H | Wagging | 333.96 | 4.4512 | 0.3 | |
H—O—Cl | Anti-Symmetric | 455.99 | 3.2037 | 0.2 | |
N—O—C | Anti-Symmetric | 563.97 | 8.6161 | 0.6 | |
Cl—N—C | Wagging | 632.99 | 13.8687 | 0.7 | |
H—O—N | Scissoring | 704.58 | 17.8445 | 1.1 | |
N—C—H | Anti-Symmetric | 761.52 | 0.5719 | 0.2 | |
N—C—Cl | Scissoring | 803.12 | 1.9640 | 0.1 | |
Cl—O—N | Scissoring | 820.29 | 2.4146 | 0.2 | |
N—N—C | Scissoring | 872.21 | 13.2304 | 0.9 | |
C—N—Cl | Anti-Symmetric | 885.06 | 7.1286 | 0.5 | |
O—N—O | Symmetric | 1097.90 | 22.6627 | 1.4 | |
N—O—N | Anti-Symmetric | 1319.69 | 3.8103 | 0.2 | |
O—N—O | Anti-Symmetric | 1350.48 | 2.8253 | 0.2 | |
N—C—N | Anti-Symmetric | 1418.41 | 1.4583 | 0.3 | |
Cl—N—N | Anti-Symmetric | 1449.32 | 2.7903 | 0.2 | |
O—N—H | Anti-Symmetric | 1613.39 | 10.2655 | 0.7 |
Compounds | EHOMO | ELUMO | Energy gap (E g) |
|---|---|---|---|
SL1 | -0.1475ev | -0.1247eV | 0.0228eV |
SL2 | -0.1329eV | -0.0724eV | 0.2063eV |
SL3 | -0.1153eV | -0.1310eV | 0.0157eV |
Energy level | Fragment 1 | Fragment 2 | Fragment 3 |
|---|---|---|---|
HOMO | 61 | 36 | 2 |
HOMO-1 | 5 | 92 | 0 |
HOMO-2 | 7 | 90 | 1 |
HOMO-3 | 54 | 91 | 0 |
HOMO-4 | 8 | 93 | 3 |
LUMO | 85 | 11 | 41 |
LUMO+1 | 57 | 3 | 19 |
LUMO+2 | 79 | 2 | 13 |
LUMO+3 | 75 | 23 | 11 |
LUMO+4 | 76 | 8 | 12 |
Energy level | Fragment 1 | Fragment 2 | Fragment 3 |
|---|---|---|---|
HOMO | 58 | 38 | 1 |
HOMO-1 | 8 | 93 | 0 |
HOMO-2 | 86 | 91 | 0 |
HOMO-3 | 57 | 46 | 1 |
HOMO-4 | 79 | 41 | 1 |
LUMO | 75 | 09 | 4 |
LUMO+1 | 80 | 1 | 54 |
LUMO+2 | 74 | 2 | 59 |
LUMO+3 | 72 | 1 | 20 |
LUMO+4 | 78 | 26 | 9 |
Energy level | Fragment 1 | Fragment 2 | Fragment 3 |
|---|---|---|---|
HOMO | 51 | 43 | 3 |
HOMO-1 | 2 | 94 | 1 |
HOMO-2 | 1 | 97 | 0 |
HOMO-3 | 4 | 92 | 1 |
HOMO-4 | 11 | 83 | 1 |
LUMO | 87 | 11 | 4 |
LUMO+1 | 89 | 0 | 17 |
LUMO+2 | 45 | 0 | 51 |
LUMO+3 | 29 | 2 | 62 |
LUMO+4 | 24 | 6 | 60 |
Donor | Type | Acceptor | Type | E (1) Kcal mol-1 | E (J)-E (I) (a. u) | F (I, J) (a. u) |
|---|---|---|---|---|---|---|
C24-C25 | Π | C23-N7 | π* | 39.48 | 0.29 | 0.107 |
C17-C18 | Π | C9-C10 | π* | 29.85 | 0.35 | 0.091 |
C15-C26 | Π | C17-C18 | π* | 24.72 | 0.37 | 0.087 |
C8-N6 | Π | C9-C10 | π* | 10.18 | 0.44 | 0.061 |
C12-C14 | Π | C1-C2 | σ * | 2.52 | 0.92 | 0.047 |
C3-O1 | Π | C18-O16 | π* | 0.93 | 0.53 | 0.021 |
C15-O2 | Π | C15-O9 | π* | 0.55 | 0.56 | 0.016 |
C18-C19 | Σ | C7-C8 | σ * | 3.98 | 1.43 | 0.068 |
C14-C18 | Σ | C8-C4 | σ * | 3.88 | 1.34 | 0.064 |
C4-C31 | Σ | C4-N5 | σ * | 3.41 | 1.36 | 0.061 |
C24-H29 | Σ | C25-C26 | σ * | 2.99 | 1.16 | 0.053 |
C13-C23 | Σ | C10-C11 | σ * | 2.74 | 1.35 | 0.054 |
C17-C18 | Σ | C18-C17 | σ * | 2.01 | 1.27 | 0.046 |
C15-C16 | Σ | C16-C17 | σ * | 1.89 | 1.29 | 0.045 |
C1-C2 | Σ | C1-N5 | σ * | 1.32 | 1.28 | 0.037 |
C17-H22 | Σ | C17-C18 | σ * | 0.84 | 1.16 | 0.028 |
C9-H13 | Σ | C8-C9 | σ * | 0.53 | 1.13 | 0.022 |
C7-O4 | Σ | C17-O3 | σ * | 0.51 | 1.62 | 0.026 |
C26 | LP(1) | C23-N2 | π* | 83.86 | 0.11 | 0.094 |
C9 | LP(1) | C15-C18 | π* | 67.27 | 0.21 | 0.128 |
O5 | LP(2) | C8-O1 | π* | 55.11 | 0.39 | 0.132 |
C18 | LP(1) | C16-C17 | π* | 47.36 | 0.2 | 0.111 |
O3 | LP(3) | C18-O4 | π* | 41.88 | 0.39 | 0.122 |
N2 | LP(1) | C11-C13 | π* | 1.03 | 0.07 | 0.031 |
O3 | LP(2) | C3-O4 | π* | 25.99 | 0.43 | 0.096 |
O4 | LP(2) | C6-C7 | σ * | 20.65 | 0.8 | 0.119 |
O2 | LP(2) | C7-O2 | σ * | 12.57 | 0.98 | 0.104 |
N2 | LP(1) | C10-C11 | σ * | 8.26 | 0.99 | 0.082 |
N5 | LP(1) | N2-H18 | σ * | 2.67 | 0.96 | 0.046 |
O1 | LP(1) | C15-O7 | σ * | 1.72 | 1.4 | 0.044 |
N2 | LP(1) | C11-C13 | σ * | 1.05 | 1.07 | 0.03 |
C8 | LP(1) | C18-N9 | σ * | 0.52 | 0.63 | 0.022 |
DFT | Density Functional Theory |
TD-DFT | Time dependent-Density Functional Theory |
DOS | Density of State |
TDM | Transition Density of Matrices |
NBO | Natural Bod Orbital |
VIE | Vertical Ionization Energy |
EDDM | Electron Density Difference Map |
FMOs | Frontier Molecular Orbitals |
MEPs | Molecular Electrostatic Potentials |
| [1] | Lasri, J., Eltayeb, N. E., Soliman, S. M., Ali, E. M., Alhayyani, S., & Akhdhar, A. (2023). Synthesis, crystal structure, DFT, and anticancer activity of some imine-type compounds via routine schiff base reaction: an example of unexpected cyclization to oxazine derivative. Molecules, 28(12), 4766. |
| [2] | Manvatkar, V. D., Patle, R. Y., Meshram, P. H., & Dongre, R. S. (2023). Azomethine-functionalized organic-inorganic framework: an overview. Chemical Papers, 77(10), 5641-5662. |
| [3] | Moanta, A. (2025). Classification, synthesis, isomerism, and spectral characterization of Schiff bases. Mini-Reviews in Organic Chemistry. |
| [4] | Mushtaq, I., Ahmad, M., Saleem, M., & Ahmed, A. (2024). Pharmaceutical significance of Schiff bases: an overview. Future Journal of Pharmaceutical Sciences, 10(1), 16. |
| [5] | Islam, M. H., & Hannan, M. A. (2024). Schiff bases: contemporary synthesis, properties, and applications. In Novelties in Schiff bases. IntechOpen. |
| [6] | Çınar, E. (2025). SCHIFF BASES: STRUCTURE, PROPERTIES AND APPLICATIONS. From Molecule to Discipline: Contemporary and Innovative Approaches in Chemical Science, 131. |
| [7] | Mariam Abdul-Bary, M. A. B., Sweah, Z. J., & Sweah, Z. J. (2026). Schiff Bases and Their Complexities: A Review. Schiff Bases and Their Complexities: A Review, 3(1), 26-48. |
| [8] | TAŞ, N. A. (2024). AMINO ACID SCHIFF BASES: DEFINITION, PROPERTIES AND APPLICATIONS. Modern Research and Investigations in Natural and Mathematical Sciences-II, 75. |
| [9] | Feng, H., Yang, P., Fu, J., Qin, M., & Zhang, K. (2025). Substituent-Engineered Multi-Stimuli-Responsive Schiff Base Crystals: from Proton Transfer Mechanisms to Smart Device Prototypes. Crystal Growth & Design, 25(19), 8203-8216. |
| [10] | Mandal, P., & Pratihar, J. L. (2023). A review of the photochromic behavior of metal complexes embedded in conjugated (-N= N-C= N-) and non-conjugated azo-imine-based ligands. Reviews in Inorganic Chemistry, 43(4), 583-625. |
| [11] | Li, D., Liang, X., Zhang, F., Li, J., Zhang, Z., Wang, S.,. & Guo, K. (2022). Imine bond orientation manipulates AIEgen derived Schiff base isomers through the intramolecular hydrogen bond effect for different fluorescence properties and applications. Journal of Materials Chemistry C, 10(30), 11016-11026. |
| [12] | Cossío, F. P., de Cózar, A., Sierra, M. A., Casarrubios, L., Muntaner, J. G., Banik, B. K., & Bandyopadhyay, D. (2022). Role of imine isomerization in the stereocontrol of the Staudinger reaction between ketenes and imines. RSC advances, 12(1), 104-117. |
| [13] | Khan, Y., Sarfraz, H., Rehman, W., Khan, M., Rasheed, L., & Rahman, K. U. (2025). Innovative horizons in drug design: exploring the synthesis and medicinal properties of heterocyclic schiff bases. A review. Mini-Reviews in Medicinal Chemistry, 25(10), 727-744. |
| [14] | Omar, I., Alharas, M. M., Feizi‐Dehnayebi, M., Alharbi, S. K., Abo‐Dief, H. M., Qasem, H. A.,. & Abu‐Dief, A. M. (2025). Design, Synthesis, Physico‐Chemical Characterization, Stability Determination, and Biomedical Applications of Some Novel Tetra‐Dentate Imine Metal Chelates Supported by Theoretical Approaches: Bridging Coordination Chemistry and Life Sciences. Applied Organometallic Chemistry, 39(3), e70056. |
| [15] | Biswas, T., Mittal, R. K., Sharma, V., Kanupriya, & Mishra, I. (2024). Schiff bases: versatile mediators of medicinal and multifunctional advancements. Letters in Organic Chemistry, 21(6), 505-519. |
| [16] | Abdelsalam, M. M., Seadawy, M. G., Eldesouky, M., & El-Sherif, A. A. (2026). Comprehensive Thesis Review Structure: Schiff Bases in Modern Medicine and Biotechnology. Egyptian Journal of Chemistry. |
| [17] | Abdel‐Rahman, L. H., Al‐Farhan, B. S., Nafady, A., Omar, I., Aljohani, F. S., Shehata, M. R., ... & Abu‐Dief, A. M. (2025). Design, Preparation, Physicochemical Characterization, and DFT Calculations of Some Novel Complexes Based on Bi‐Dentate Imine Ligand: Biomedical Applications and Molecular Docking Approach. Applied Organometallic Chemistry, 39(3), e70052. |
| [18] | Qian, C., Feng, L., Teo, W. L., Liu, J., Zhou, W., Wang, D., & Zhao, Y. (2022). Imine and imine-derived linkages in two-dimensional covalent organic frameworks. Nature Reviews Chemistry, 6(12), 881-898. |
| [19] | Rudolf, R., Neuman, N. I., Walter, R. R., Ringenberg, M. R., & Sarkar, B. (2022). Mesoionic Imines (MIIs): Strong Donors and Versatile Ligands for Transition Metals and Main Group Substrates. Angewandte Chemie International Edition, 61(25), e202200653. |
| [20] | Elangovan, N., Sowrirajan, S., Alzahrani, A. Y. A., Rajendran Nair, D. S., & Thomas, R. (2024). Fluorescent azomethine by the condensation of sulfadiazine and 4-chlorobenzaldehyde in solution: synthesis, characterization, solvent interactions, electronic structure, and biological activity prediction. Polycyclic Aromatic Compounds, 44(4), 2332-2353. |
| [21] | Bakr, E. A., Atteya, E. H., Al‐Hefnawy, G. B., El‐Attar, H. G., & El‐Gamil, M. M. (2023). A novel azo‐azomethine benzoxazole‐based ligand and its transition metal (II), (III), (IV) complexes: Synthesis, characterization, theoretical studies, biological evaluation, and catalytic application. Applied Organometallic Chemistry, 37(4), e7042. |
| [22] | Dömötör, O., May, N. V., Gál, G. T., Spengler, G., Dobrova, A., Arion, V. B., & Enyedy, É. A. (2022). Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity. Molecules, 27(7), 2044. |
| [23] | Liu, Y., Bian, C., Li, Y., Sun, P., Xiao, Y., Xiao, X.,. & Dong, X. (2023). Aminobenzaldehyde convelently modified graphitic carbon nitride photocatalyst through Schiff base reaction: Regulating electronic structure and improving visible-light-driven photocatalytic activity for moxifloxacin degradation. Journal of Colloid and Interface Science, 630, 867-878. |
| [24] | Raczuk, E., Dmochowska, B., Samaszko-Fiertek, J., & Madaj, J. (2022). Different Schiff bases—structure, importance and classification. Molecules, 27(3), 787. |
| [25] | El Batouti, M., El-Mossalamy, E. H., & Elewa, M. M. (2026). Donor-acceptor dichotomy in novel Schiff bases: comprehensive spectroscopic and DFT investigation of intramolecular hydrogen bonding and charge-transfer properties. RSC advances, 16(22), 19729-19742. |
| [26] | Sánchez-Pacheco, A. D., Hernández-Vergara, M., Jaime-Adán, E., Hernández-Ortega, S., & Valdés-Martínez, J. (2021). Schiff bases as possible hydrogen bond donors and acceptors. Journal of Molecular Structure, 1234, 130136. |
| [27] | Nath, S., Bhattacharya, B., Sarkar, U., & Singh, T. S. (2022). Solvent effects on the photophysical properties of a donor-acceptor based Schiff base. Journal of Fluorescence, 32(4), 1321-1336. |
| [28] | Er-rajy, M., Salghi, R., Elhallaoui, M., Azzaoui, K., Chafiq, M., Elboughdiri, N.,. & Ko, Y. G. (2025). Exploring donor-acceptor characteristics and adsorption behavior of a naphthamide-based inhibitor for protective surfaces through a molecular modeling approach. Journal of the Indian Chemical Society, 102(4), 101640. |
| [29] | Lakhera, S., Rana, M., & Devlal, K. (2022). Theoretical study on spectral and optical properties of essential amino acids: a comparative study. Optical and Quantum Electronics, 54(11), 714. |
| [30] | Alkhatib, F. M., & Alsulami, H. M. (2023). Synthesis, characterization, DFT calculations and biological activity of new Schiff base complexes. Heliyon, 9(8). |
| [31] | Yavuz, Ş., Çolak, N., Yıldırım, T., Köse, D. A., & Şahin, O. (2026). Complexes formed by a heterocyclic schiff base containing an aminothiophene group with Ni (II) and Co (II) metals and their structural characterization. Journal of Molecular Structure, 1357, 145217. |
| [32] | Raju, S. K., Settu, A., Thiyagarajan, A., Rama, D., Sekar, P., & Kumar, S. (2022). Biological applications of Schiff bases: An overview. GSC Biol. Pharm. Sci, 21(3), 203-215. |
| [33] | Wang, L., & Yang, D. (2023). Take Advantage of the N‐Nucleophilicity of Imine in Catalytic Cyclization Reactions. ChemCatChem, 15(9), e202300189. |
| [34] | Martínez, R. F., Matamoros, E., Cintas, P., & Palacios, J. C. (2020). Imine or Enamine? Insights and predictive guidelines from the electronic effect of substituents in H-bonded salicylimines. The Journal of Organic Chemistry, 85(9), 5838-5862. |
| [35] | Lv, H., Du, Y., Zhang, H., Zheng, Y., Yan, Z., & Dong, N. (2023). Advances in Mannich‐type Reactions Based on the Classification of Compounds with Activated α‐H. ChemistrySelect, 8(21), e202300173. |
| [36] | Guan, H., Sun, H., & Zhao, X. (2025). Application of density functional theory to molecular engineering of pharmaceutical formulations. International journal of molecular sciences, 26(7), 3262. |
| [37] | Tegegn, D. F., Belachew, H. Z., & Salau, A. O. (2024). DFT/TDDFT calculations of geometry optimization, electronic structure and spectral properties of clevudine and telbivudine for treatment of chronic hepatitis B. Scientific Reports, 14(1), 8146. |
| [38] | Napiórkowska, E., Milcarz, K., & Szeleszczuk, Ł. (2023). Review of applications of density functional theory (DFT) quantum mechanical calculations to study the high-pressure polymorphs of organic crystalline materials. International Journal of Molecular Sciences, 24(18), 14155. |
| [39] | Ahmed, L., & Omer, R. (2020). Population Analysis and UV-Vis spectra of Dopamine Molecule Using Gaussian 09. Journal of Physical Chemistry and Functional Materials, 3(2), 48-58. |
| [40] | Bálint, D., & Jäntschi, L. (2021). Comparison of molecular geometry optimization methods based on molecular descriptors. Mathematics, 9(22), 2855. |
| [41] | Kanagathara, N., & Nanmaran, R. (2021). Illustration of potential energy surface from DFT calculation along with fuzzy logic modelling for optimization of N-acetylglycine. Computational and Theoretical Chemistry, 1202, 113301. |
| [42] | Nkoe, P., Manicum, A. L. E., Louis, H., Malan, F. P., Nzondomyo, W. J., Chukwuemeka, K.,. & Unimuke, T. O. (2023). Influence of solvation on the spectral, molecular structure, and antileukemic activity of 1-benzyl-3-hydroxy-2-methylpyridin-4(1H)-one. Journal of Molecular Liquids, 370, 121045. |
| [43] | Limbu, S., Ojha, T., Ghimire, R. R., & Rai, K. B. (2024). An investigation of vibrational analysis, thermodynamics properties and electronic properties of Formaldehyde and its stretch by substituent acetone, acetyl chloride and methyl acetate using first principles analysis. BIBECHANA, 21(1), 23-36. |
| [44] | Gounhalli, S. G., Bhagyalaxmi, B., Konda, R. B., & Shivaleela, B. (2024). DFT Studies on Excited State Geometry, Vibrational Modes, NMR, Molecular Orbital and Mulliken Charges of Laser Dye. Mapana Journal of Sciences, 23(4), 53. |
| [45] | Wu, T., Fang, Z., Wang, Z., Liu, L. E., Song, J., & Song, J. (2023). Stability, electronic and catalytic properties of ConMoP (n= 1~ 5) clusters: A DFT study. Journal of molecular modeling, 29(8), 269. |
| [46] | Yildiz, E. A., Pepe, Y., Erdener, D., Karatay, A., Boyacioglu, B., Ünver, H.,. & Elmalı, A. (2023). Effect of group electronegativity on spectroscopic, biological, chromogenic sensing and optical properties of 2-formyl-benzene sulfonic acid sodium salt-based Schiff bases. Journal of Molecular Structure, 1286, 135611. |
| [47] | Li, L., Xu, S., Jin, L., Chen, W., & Chen, S. (2025). Efficient synergistic enhancing degradation of waste poly (ethylene terephthalate) by combination of DBU with zinc 2-ethylhexanoate. Journal of Environmental Chemical Engineering, 13(3), 116308. |
| [48] | Asrafali, S. P., Periyasamy, T., & Kim, S. C. (2023). Rapid transformation in wetting properties of PTFE membrane using plasma treatment. Polymers, 15(19), 3874. |
| [49] | Kahraman, S., Hepokur, C., Erci, F., Erkan, S., Cetin, S., Kose, M., & Kurtoglu, M. (2025). Copper (II) complexes with N, O-donor azo-Schiff base ligands: Synthesis, structure, DFT studies, molecular docking, anticancer and antimicrobial activity. Polyhedron, 269, 117393. |
| [50] | Liao, G., Ruan, M., Wang, Y., Chen, H., & Weng, Y. (2025). IR fingerprint of the intermolecular hydrogen bond on amino acids and its relevance to chaperone activity of αB-crystallin. The Journal of Physical Chemistry B, 129(4), 1237-1247. |
| [51] | Xiao, P., Liu, S., Zhou, X., Huang, E., Zhong, L., Zhang, W.,. & Dong, W. (2024). Structure and vibrational spectroscopy of 2-methylallyl alcohol. Chinese Journal of Chemical Physics, 37(4), 481-489. |
| [52] | Ali, H. F., Adeel, M., Aiman, U., Jamal, S., Haroon, M., & Ahamad, T. (2025). Synthesis, spectral characterization and nonlinear optical exploration of potent fluorene-based compounds: A DFT refine experimental study. Journal of Molecular Structure, 143630. |
| [53] | Shafiq, I., Irshad, I., Zahid, R., Mahmood, K., Ahmed, S., Bullo, S., & Alhokbany, N. (2025). Exploration of promising key electronic and nonlinear optical properties of bifluorenylidene based chromophores: a TD-DFT/DFT approach. Scientific Reports, 15(1), 10701. |
| [54] | Khalid, M., Khan, M., Gull, K., Mubarak, M., Arshad, M. N., & Imran, M. (2026). Exploring the NLO potential of new designed thieno[2]indole-based derivatives via donor group modification: a DFT study on static and frequency-dependent NLO properties. Journal of Computational Electronics, 25(1), 46. |
| [55] | Lu, T. (2025). Visualization analysis of covalent and noncovalent interactions in real space. Angewandte Chemie International Edition, 64(29), e202504895. |
| [56] | Lu, T. (2024). A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. The Journal of chemical physics, 161(8). |
| [57] | Marimuthu, M., Balasubramanian, S., Viswanathan, R. B., Parthiban, H. P., Mahalingam, M., & Kasirajan, G. (2025). An in-silico investigation of 8-hydroxyquinoline (8-HoQ) derivatives as potential anti-tuberculosis agents by targeting glutamate kinase. Structural Chemistry, 1-21. |
| [58] | Warburton, R. E., Soudackov, A. V., & Hammes-Schiffer, S. (2022). Theoretical modeling of electrochemical proton-coupled electron transfer. Chemical reviews, 122(12), 10599-10650. |
| [59] | Fichthorn, K. A., Theory of anisotropic metal nanostructures. Chemical Reviews, 2023. 123(7): p. 4146-4183. |
| [60] | Sangari, S., Lackmy-Vallee, A., Preuilh, A., Peyre, I., Pradat, P. F., & Marchand-Pauvert, V. (2024). Synaptic dynamics linked to widespread elevation of H-reflex before peripheral denervation in amyotrophic lateral sclerosis. Journal of Neurophysiology, 132(5), 1541-1560. |
| [61] | Allison, D., & Wong, M. (2026). Posterior Fossa. In Operative Neurophysiology: Practical Application for Comprehensive Neuromonitoring and Neuromapping Techniques (pp. 431-466). Cham: Springer Nature Switzerland. |
| [62] | Toriyama, M. Y., Ganose, A. M., Dylla, M., Anand, S., Park, J., Brod, M. K.,. & Snyder, G. J. (2022). How to analyse a density of states. Materials today electronics, 1, 100002. |
| [63] | Fung, V., Hu, G., Ganesh, P., & Sumpter, B. G. (2021). Machine learned features from density of states for accurate adsorption energy prediction. Nature communications, 12(1), 88. |
| [64] | Sun, H. (2024). Unraveling the structure-property relationship of novel thiophene and furan‐fused cyclopentadienyl chromophores for nonlinear optical applications. Journal of Computational Chemistry, 45(31), 2612-2623. |
| [65] | UrRehman, S., Anwer, M., BiBi, S., Jamil, S., Yasin, M., Khan, S. R.,. & Jia, R. (2022). DFT analysis of different substitutions on optoelectronic properties of carbazole-based small acceptor materials for Organic Photovoltaics. Materials Science in Semiconductor Processing, 140, 106381. |
| [66] | Contreras, P., Seijas, L., & Osorio, D. (2021). TDOS quantum mechanical visual analysis for single molecules. arXiv preprint arXiv: 2105.12830. |
| [67] | Louis, H., Ifediora, L. P., Enudi, O. C., Unimuke, T. O., Asogwa, F. C., & Moshood, Y. L. (2021). Evaluation of the excited state dynamics, photophysical properties, and the influence of donor substitution in a donor-π-acceptor system. Journal of molecular modeling, 27(10), 284. |
| [68] | Ziadi, K., Aouragh, A., & Messaoudi, A. (2025). Structure-property analysis of dithienopyrrole-based D-π-A-π-D compounds: electronic and nonlinear optical responses with advanced python-based visualizations. Journal of Molecular Graphics and Modelling, 140, 109113. |
| [69] | Jamal, S., Raza, N., Khalid, M., & Braga, A. A. C. (2025). Unveiling electronic and remarkable non-linear optical properties of boron-nitrogen carbazole-based compounds via modification of π-linker and donor units: a DFT study. RSC advances, 15(11), 8262-8274. |
| [70] | Li, N., Zhang, L., & Wang, J. (2023). Modulation of chiral spectral deflection by van der Waals force-induced molecular electropolarization in catenane oligomers. RSC advances, 13(16), 11055-11061. |
| [71] | Khalid, M., Ahmed, R., Shafiq, I., Arshad, M., Asghar, M. A., Munawar, K. S.,. & Braga, A. A. (2022). First theoretical framework for highly efficient photovoltaic parameters by structural modification with benzothiophene-incorporated acceptors in dithiophene based chromophores. Scientific Reports, 12(1), 20148. |
| [72] | Ren, Y., Gai, X., Wang, J., & Wang, L. (2025). Physical mechanism of nonlinear optical properties in Lemniscular carbon nanohoops. Chemical Physics, 593, 112655. |
| [73] | Al-Matar, H. M., BinSabt, M. H., & Shalaby, M. A. (2025). Synthesis, Photophysical, and computational investigation of poly substituted pyridines. Journal of Molecular Structure, 143298. |
| [74] | Sultan, N., Ikreedeegh, R. R., & Janjua, M. R. S. A. (2026). Review and Insights into the Impact of Transition Density Matrix (TDM) on the Optoelectronic Properties of Organic Solar Cells (OSCs). High Energy Chemistry, 60(1), 1-15. |
| [75] | Aldawsari, F. S., Albugami, F. S., Altalhi, T. A., Refat, M. S., Shakya, S., Adam, A. M. A.,. & Jaber Altalhi, A. A. (2025). SPECTROSCOPIC MEASUREMENTS, COMPUTATIONAL CALCULATIONS, AND DRUG ASSAY APPLICATIONS OF CHARGE-TRANSFER COMPLEXES COMPRISING THE PHARMACEUTICAL TRIAMTERENE AND THE ORGANIC ACCEPTORS TCNQ AND DDQ. Bulletin of the Chemical Society of Ethiopia, 39(10). |
| [76] | Shafiq, I., Khalid, M., Asghar, M. A., Baby, R., Braga, A. A., Alshehri, S. M., & Ahmed, S. (2023). Influence of azacycle donor moieties on the photovoltaic properties of benzo [c, 1, 2, 5] thiadiazole based organic systems: a DFT study. Scientific Reports, 13(1), 14630. |
| [77] | Laplaza, R., Peccati, F., A. Boto, R., Quan, C., Carbone, A., Piquemal, J. P.,. & Contreras‐García, J. (2021). NCIPLOT and the analysis of noncovalent interactions using the reduced density gradient. Wiley Interdisciplinary Reviews: Computational Molecular Science, 11(2), e1497. |
| [78] | Morales‐Pumarino, D., & Barquera‐Lozada, J. E. (2023). Electron density and its reduced density gradient in the study of π-π interactions. International Journal of Quantum Chemistry, 123(18), e27051. |
| [79] | Guerra, C., Burgos, J., Ayarde-Henríquez, L., & Chamorro, E. (2024). Formulating reduced density gradient approaches for noncovalent interactions. The Journal of Physical Chemistry A, 128(30), 6158-6166. |
| [80] | Zhang, H., Liu, S., You, J., Liu, C., Zheng, S., Lu, Z.,. & Shao, B. (2024). Overcoming the barrier of orbital-free density functional theory for molecular systems using deep learning. Nature Computational Science, 4(3), 210-223. |
| [81] | Lu, T., & Chen, Q. (2022). Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. Journal of computational chemistry, 43(8), 539-555. |
| [82] | Kalita, B., Li, L., McCarty, R. J., & Burke, K. (2021). Learning to approximate density functionals. Accounts of Chemical Research, 54(4), 818-826. |
| [83] | Li Manni, G., Fdez. Galván, I., Alavi, A., Aleotti, F., Aquilante, F., Autschbach, J.,. & Lindh, R. (2023). The OpenMolcas Web: A community-driven approach to advancing computational chemistry. Journal of Chemical Theory and Computation, 19(20), 6933-6991. |
| [84] | Choudhary, K., DeCost, B., Chen, C., Jain, A., Tavazza, F., Cohn, R.,. & Wolverton, C. (2022). Recent advances and applications of deep learning methods in materials science. npj Computational Materials, 8(1), 59. |
| [85] | Agwupuye, J. A., Louis, H., Unimuke, T. O., David, P., Ubana, E. I., & Moshood, Y. L. (2021). Electronic structure investigation of the stability, reactivity, NBO analysis, thermodynamics, and the nature of the interactions in methyl-substituted imidazolium-based ionic liquids. Journal of Molecular Liquids, 337, 116458. |
| [86] | Jawad, H. M., & Jasim, F. A. (2024, May). Theoretical investigations on the natural bond orbital, HOMO-LUMO, contour maps, and energy gap of diatrizoate. In AIP conference proceedings (Vol. 3097, No. 1, p. 090004). AIP Publishing LLC. |
| [87] | Santos‐Jr, C. V., Kraka, E., & Moura Jr, R. T. (2025). Chemical bond overlap descriptors from multiconfiguration wavefunctions. Journal of Computational Chemistry, 46(1), e27534. |
| [88] | Edache, E. I., Uzairu, A., Mamza, P. A., Shallangwa, G. A., & Ibrahim, M. T. (2024). DFT studies on structure, electronics, bonding nature, NBO analysis, thermodynamic properties, molecular docking, and MM-GBSA evaluation of 4-methyl-3- [2-(4-nitrophenyl)-1, 3-dioxo-2, 3-dihydro-1 H-isoindole-5-amido] benzoic acid: a potent inhibitor of Graves’ disease. Journal of Umm Al-Qura University for Applied Sciences, 10(4), 652-670. |
| [89] | Miller, S. A. (2023). The location of the chemical bond. Application of long covalent bond theory to the structure of silica. Frontiers in chemistry, 11, 1123322. |
| [90] | Weinhold, F. (2023). “Noncovalent interaction”: A chemical misnomer that inhibits proper understanding of hydrogen bonding, rotation barriers, and other topics. Molecules, 28(9), 3776. |
| [91] | Prabakaran, A., Maheswari, C. U., Issaoui, N., Al-Dossary, O. M., Rajamani, T., Gnanasambandan, T.,. & Manikandan, A. (2024). Computational insight into the spectroscopic and molecular docking analysis of estrogen receptor with ligand 2, 3-dimethyl-N [2-(hydroxy) benzylidene] aniline. Journal of King Saud University-Science, 36(6), 103196. |
| [92] | Shiny, C. L., Rathika, A., Tarika, J. D., Alen, S., & Beaula, T. J. (2023). Effect of charge transfer and non-covalent interactions of the synthesized NLO compound p-nitrophenol sodium-bisulfate using DFT method. Polycyclic Aromatic Compounds, 43(5), 4621-4639. |
| [93] | Medimagh, M., Issaoui, N., Gatfaoui, S., Kazachenko, A. S., Al-Dossary, O. M., Kumar, N.,. & Bousiakoug, L. G. (2023). Investigations on the non-covalent interactions, drug-likeness, molecular docking and chemical properties of 1, 1, 4, 7, 7-pentamethyldiethylenetriammonium trinitrate by density-functional theory. Journal of King Saud University-Science, 35(4), 102645. |
APA Style
Javid, M., Shahzadi, I., Jamil, F., Asghar, S., Abbas, M. S., et al. (2026). A Computational Investigation of Spectroscopic, Molecular, and Electrostatic Properties of the Schiff Base Molecules via DFT. International Journal of Computational and Theoretical Chemistry, 14(1), 15-33. https://doi.org/10.11648/j.ijctc.20261401.12
ACS Style
Javid, M.; Shahzadi, I.; Jamil, F.; Asghar, S.; Abbas, M. S., et al. A Computational Investigation of Spectroscopic, Molecular, and Electrostatic Properties of the Schiff Base Molecules via DFT. Int. J. Comput. Theor. Chem. 2026, 14(1), 15-33. doi: 10.11648/j.ijctc.20261401.12
@article{10.11648/j.ijctc.20261401.12,
author = {Muhammad Javid and Ifra Shahzadi and Farah Jamil and Sabahat Asghar and Muhammad Sajid Abbas and Ihsan Maseeh and Muhammad Hasnain and Muhammad Zohaib Sabir},
title = {A Computational Investigation of Spectroscopic, Molecular, and Electrostatic Properties of the Schiff Base Molecules via DFT},
journal = {International Journal of Computational and Theoretical Chemistry},
volume = {14},
number = {1},
pages = {15-33},
doi = {10.11648/j.ijctc.20261401.12},
url = {https://doi.org/10.11648/j.ijctc.20261401.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijctc.20261401.12},
abstract = {This theoretical work give a comprehensive properties of a Schiff base compound that is formed when amines and ketone or aldehyde are combined. The investigated Schiff base compounds were designed by combining primary amines with ketones or aldehydes. The spectroscopic characteristics, molecular structure, electrostatic potential maps, and other molecular properties of these compounds had been computed at the B3LYP Functional with a 6-311G(d,p) basic set. The optimization and transition states of the molecules were analyzed by applying the B3LYP Functional with a 6-31G(d,p) basic set, based on density functional theory (DFT) and time-dependent density functional theory (TD-DFT) for ground-state and excited-state calculations, respectively. We had also determine the band length and band angles of the designed molecules. Several computational software packages was used to study the spectroscopic, electronics, and molecular characteristics of explore Schiff base compounds. The molecules were designed in GaussView5 and optimized in Gaussian 09. PyMolyze and Origin6.0 software were used to perform the density of state (DOS) analysis & to draw the absorption spectra of probe molecules. TDM analysis were conducted to determine the charge distribution in the investigated molecules using Multiwfn3.7 and VMD1.9.1 software. Correlation statistical models were employed to interpret the statistical data. The docking results of the designed molecules were compared with antibacterial standards, and we expect these results to show greater efficiency than the reference molecules. Additionally, the antitumor and antibacterial characteristics of a designed molecule were compared with those of the reference molecules.},
year = {2026}
}
TY - JOUR T1 - A Computational Investigation of Spectroscopic, Molecular, and Electrostatic Properties of the Schiff Base Molecules via DFT AU - Muhammad Javid AU - Ifra Shahzadi AU - Farah Jamil AU - Sabahat Asghar AU - Muhammad Sajid Abbas AU - Ihsan Maseeh AU - Muhammad Hasnain AU - Muhammad Zohaib Sabir Y1 - 2026/07/08 PY - 2026 N1 - https://doi.org/10.11648/j.ijctc.20261401.12 DO - 10.11648/j.ijctc.20261401.12 T2 - International Journal of Computational and Theoretical Chemistry JF - International Journal of Computational and Theoretical Chemistry JO - International Journal of Computational and Theoretical Chemistry SP - 15 EP - 33 PB - Science Publishing Group SN - 2376-7308 UR - https://doi.org/10.11648/j.ijctc.20261401.12 AB - This theoretical work give a comprehensive properties of a Schiff base compound that is formed when amines and ketone or aldehyde are combined. The investigated Schiff base compounds were designed by combining primary amines with ketones or aldehydes. The spectroscopic characteristics, molecular structure, electrostatic potential maps, and other molecular properties of these compounds had been computed at the B3LYP Functional with a 6-311G(d,p) basic set. The optimization and transition states of the molecules were analyzed by applying the B3LYP Functional with a 6-31G(d,p) basic set, based on density functional theory (DFT) and time-dependent density functional theory (TD-DFT) for ground-state and excited-state calculations, respectively. We had also determine the band length and band angles of the designed molecules. Several computational software packages was used to study the spectroscopic, electronics, and molecular characteristics of explore Schiff base compounds. The molecules were designed in GaussView5 and optimized in Gaussian 09. PyMolyze and Origin6.0 software were used to perform the density of state (DOS) analysis & to draw the absorption spectra of probe molecules. TDM analysis were conducted to determine the charge distribution in the investigated molecules using Multiwfn3.7 and VMD1.9.1 software. Correlation statistical models were employed to interpret the statistical data. The docking results of the designed molecules were compared with antibacterial standards, and we expect these results to show greater efficiency than the reference molecules. Additionally, the antitumor and antibacterial characteristics of a designed molecule were compared with those of the reference molecules. VL - 14 IS - 1 ER -
Department of Chemistry, University of Agriculture Faisalabad, Punjab, Pakistan
Institute of Chemistry, Khwaja Fareed University of Engineering and Information Technology, Rahim Yar Khan, Punjab, Pakistan
Department of Applied Chemistry, Government College University Faisalabad, Punjab, Pakistan
Institute of Chemistry, Khwaja Fareed University of Engineering and Information Technology, Rahim Yar Khan, Punjab, Pakistan
Department of Physics, University of Agriculture Faisalabad, Punjab, Pakistan
Institute of Chemistry, The Islamia University of Bahawalpur, Punjab, Pakistan
Department of Chemistry, University of Agriculture Faisalabad, Punjab, Pakistan
Department of Chemistry, King Mongkut's Institute of Technology Ladkrabang, Bangkok, Thailand
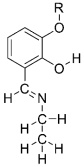
Figure 1. Designed Schiff base investigated molecule.
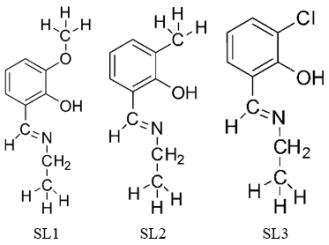
Figure 2. (SL1) 2-((ethylamino) methyl)-6-methoxyphenol, (SL2) 2-((ethylamino) methyl)-6-methylphenol, (SL3) 2-((ethylamino) methyl)-6-chlorophenol.
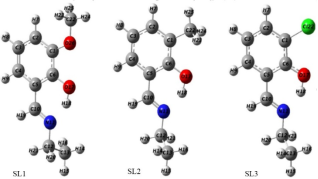
Figure 3. Ground state optimized structures of designed molecules.
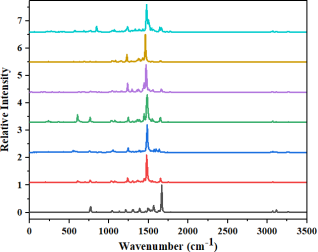
Figure 4. IR Spectrum of SL1, SL2, and SL3.
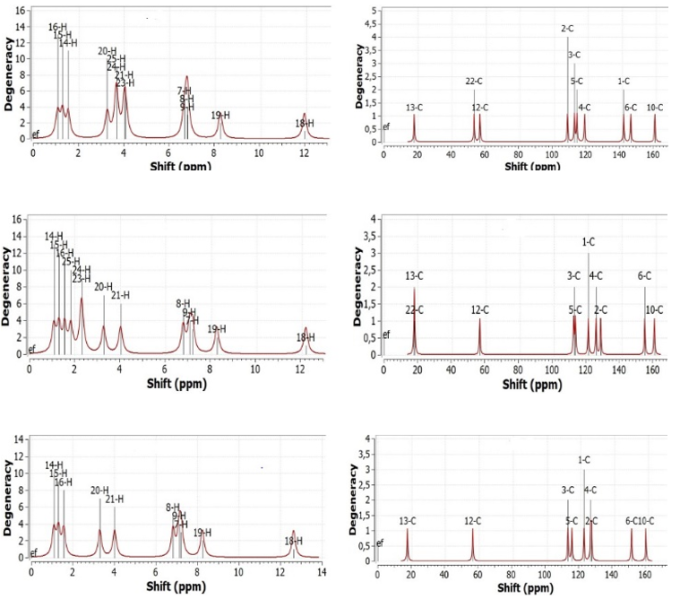
Figure 5. NMR peaks of SL1, SL2, and SL3.
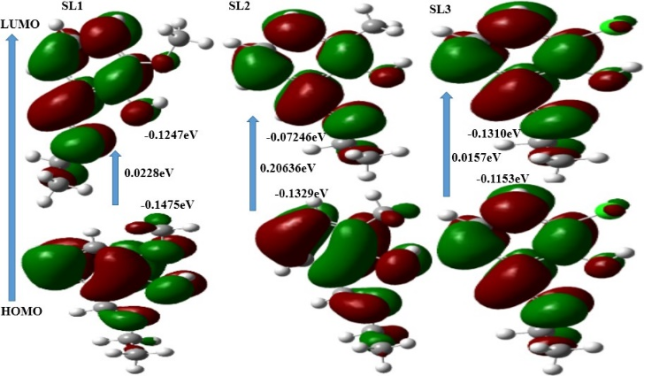
Figure 6. HOMO and LUMO of SL1, SL2, and SL3 Molecule.
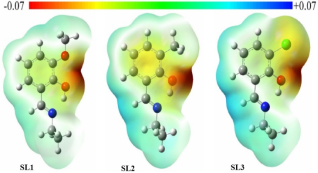
Figure 7. Electrostatic potential maps of the designed molecules.
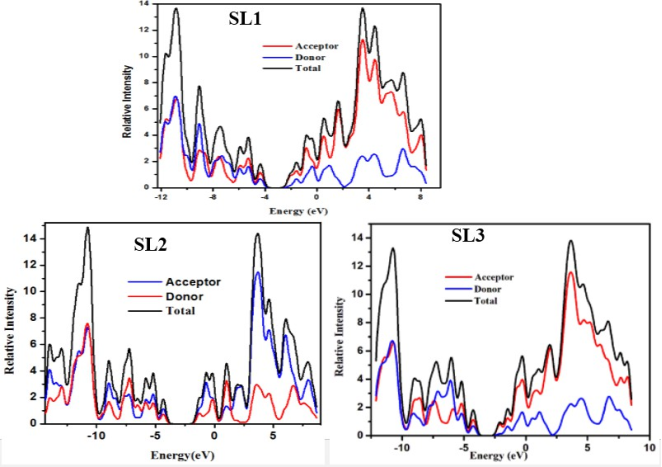
Figure 8. Density of state of SL1, SL2, and SL3 molecules.
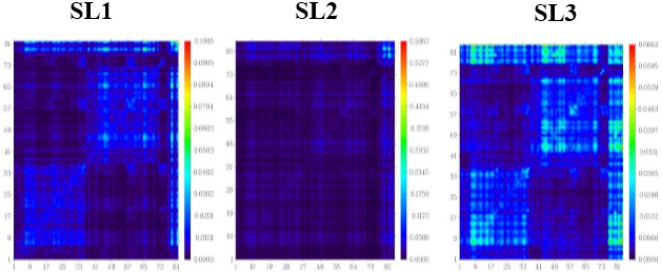
Figure 9. Two-dimensional graph illustrating the total density matrix (TDM) for the studied molecules (SL1, SL2, and SL3) is presented utilizing the DFT/CAM-Bee-3LYPmethod, with a specific emphasis on the A-D-A molecular configuration.
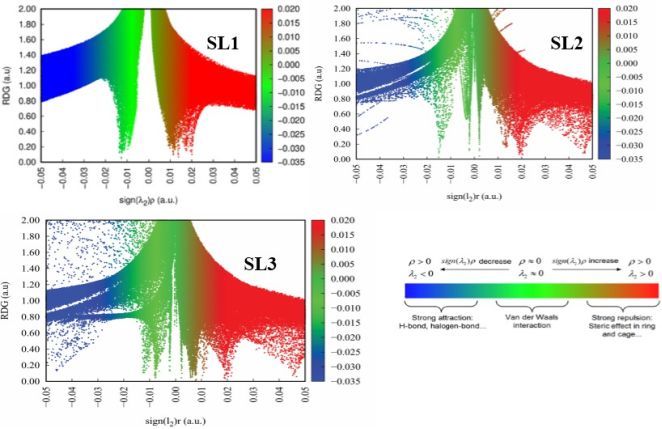
Figure 10. RDG values of SL1, SL2, and SL3.
Information

